NMR Prediction (Quick)
Details
Relatively good and fast NMR predictions can be obtained by using GFN2-xTB for
geometries (with xtb) and PBEh-3c for NMR predictions (with ORCA). For NMR
predictions, the method used for the geometry does not have to match with the method used
for the NMR prediction (unlike frequency calculations). In fact, it is common to optimize
the geometries in a vacuum using a modest basis set (ex. 6-31+G(d,p)) and
predict the NMR shifts with solvation using a large basis set (ex.
6-311++G(d,p)). The calculated NMR shifts do not correspond directly to the
experimental shifts, but are linearly correlated to them. To obtain predicted shifts (in
other words, shifts that should be close to the experimental shifts), the calculated values
must be corrected using reference compounds. This could be done with a single compound (ex.
TMS), but the fit will be better if several compounds are used. In addition to this, the
slope between calculated shifts and experimental shifts may not be exactly $1.00$; having a
range of reference compounds allows to correct this deviation.
To benchmark this method, a
set of 77 small organic molecules was used (Prepared by Lodewyk, Siebert, and Tantillo.) Three of the compounds in the original set were ignored due to the importance of
relativistic effects (which are not taken in consideration by the method.) These effect
arise with heavy atoms, such as multiple chlorine atoms bound to the same carbon. This
effect should be negligible for most molecules, although it is worth keeping in mind. As the
experimental NMR chemical shifts were measured in chloroform, this solvent was used in both
computational models. The shifts of equivalent atoms were averaged. Since the reference
molecules are small and rigid, no conformational search was necessary. The final method is
PBEh-3c (SMD, CHCl3) // GFN2-xTB (GBSA, CHCl3)
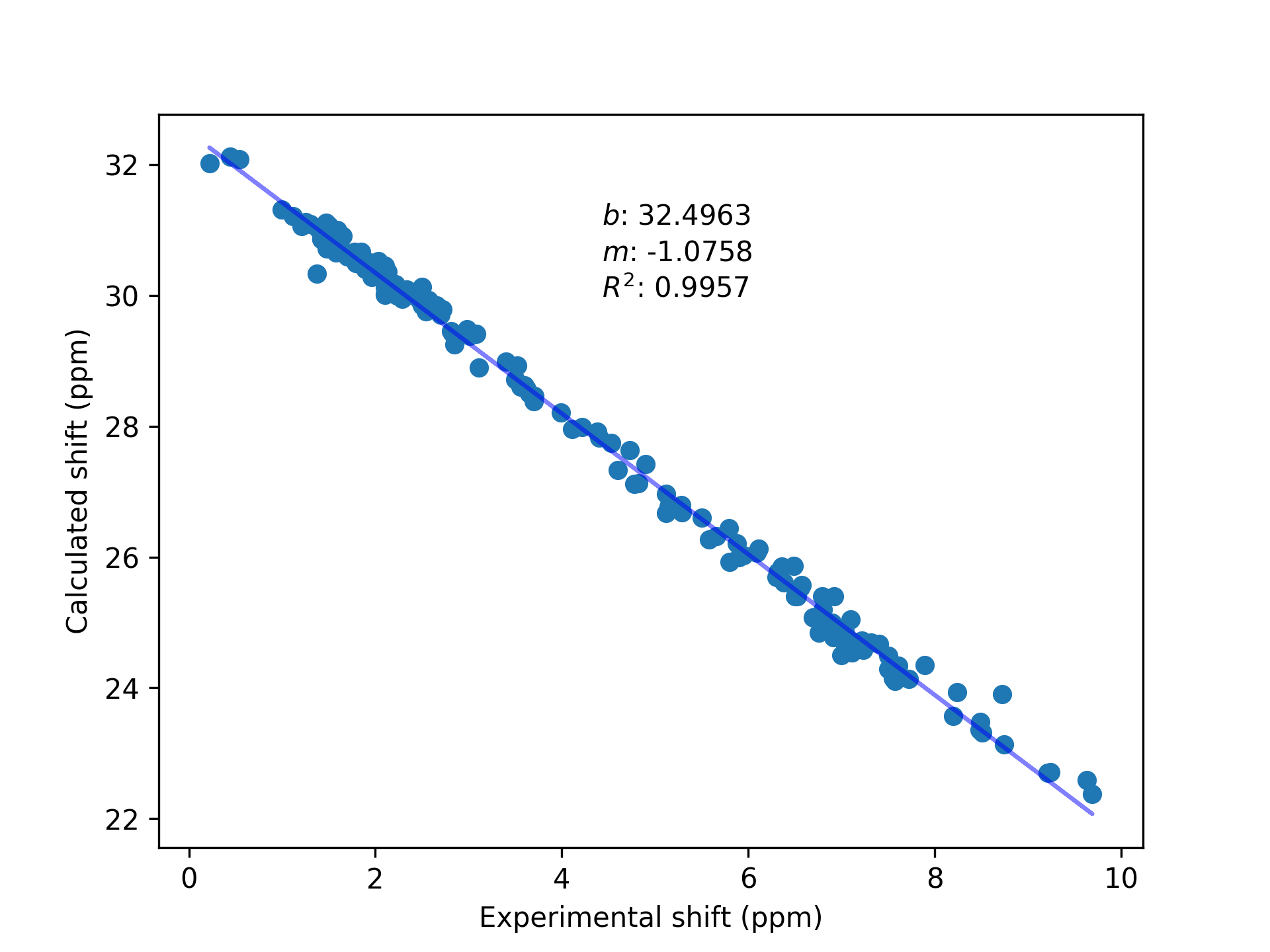
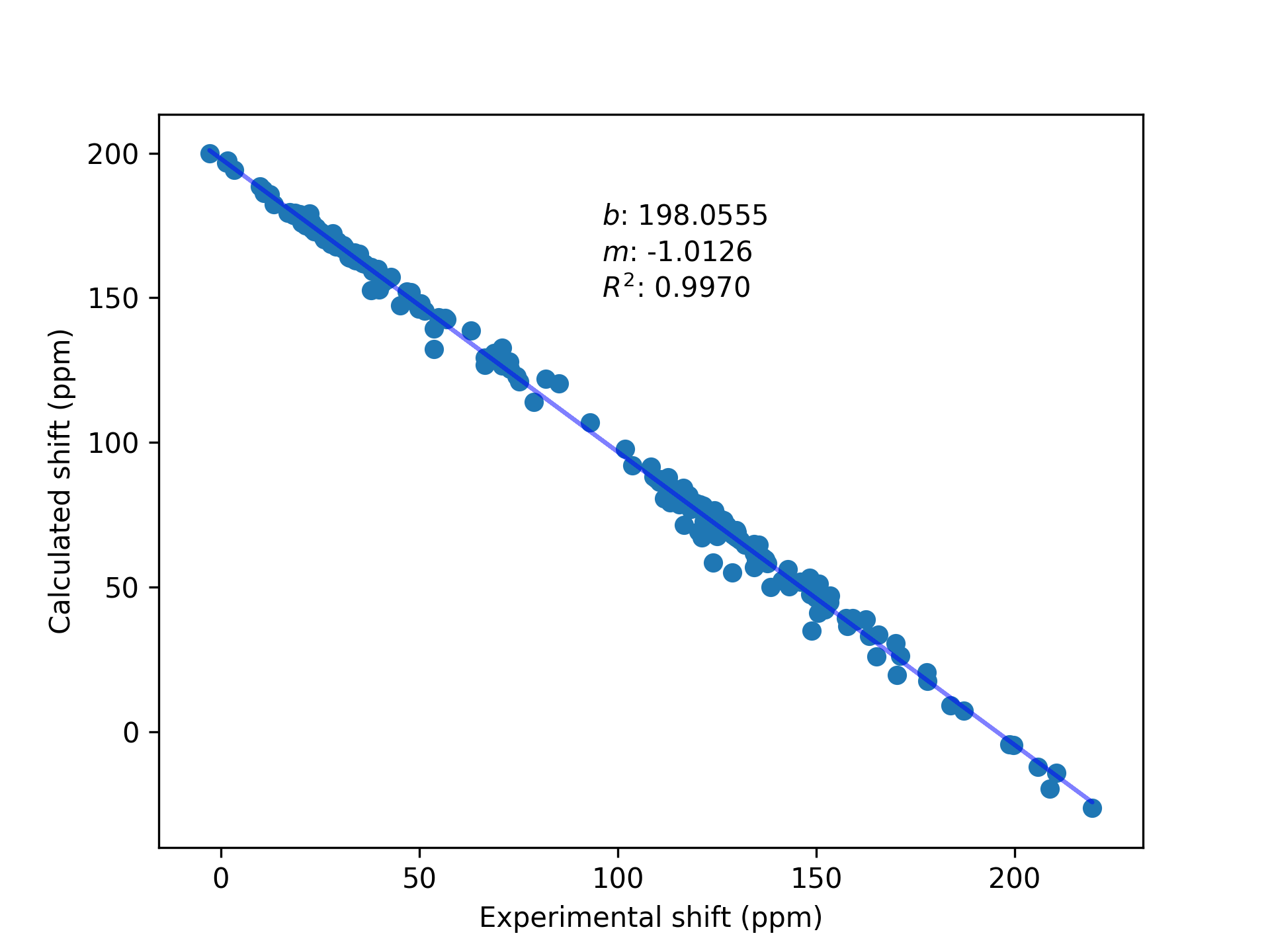
CalcUS will automatically apply the regression (if this exact method is used) and calculate the Boltzmann averaged chemical shifts. These results will be shown in the "Ensemble Properties" tab on the ensemble page. The chemical shifts of individual structures can be inspected below, in the "NMR" section.
Recipe
-
Optimize your structure with
GFN2-xTB(default setting for xtb) and solvation in chloroform (GBSA). If the molecule is flexible, perform a conformational search with the same settings. -
Perform an NMR prediction on the resulting ensemble using
PBEh-3cwith ORCA and SMD solvation in chloroform. Specify the additional keywordDef2/JK. You can filter the ensemble to perform the NMR prediction only on the most stable conformers (above 0.01 or 0.02 of Boltzmann weight is a good rule of thumb.) - If your compound has equivalent atoms, average their chemical shifts.